

Enhancing Clinical Trial Selection for Cancer Patients Using Large Language Models
Abstract
Purpose:
Methods:
Results:
Conclusion:
Introduction
Natural Language Processing
Large Language Models
Methods
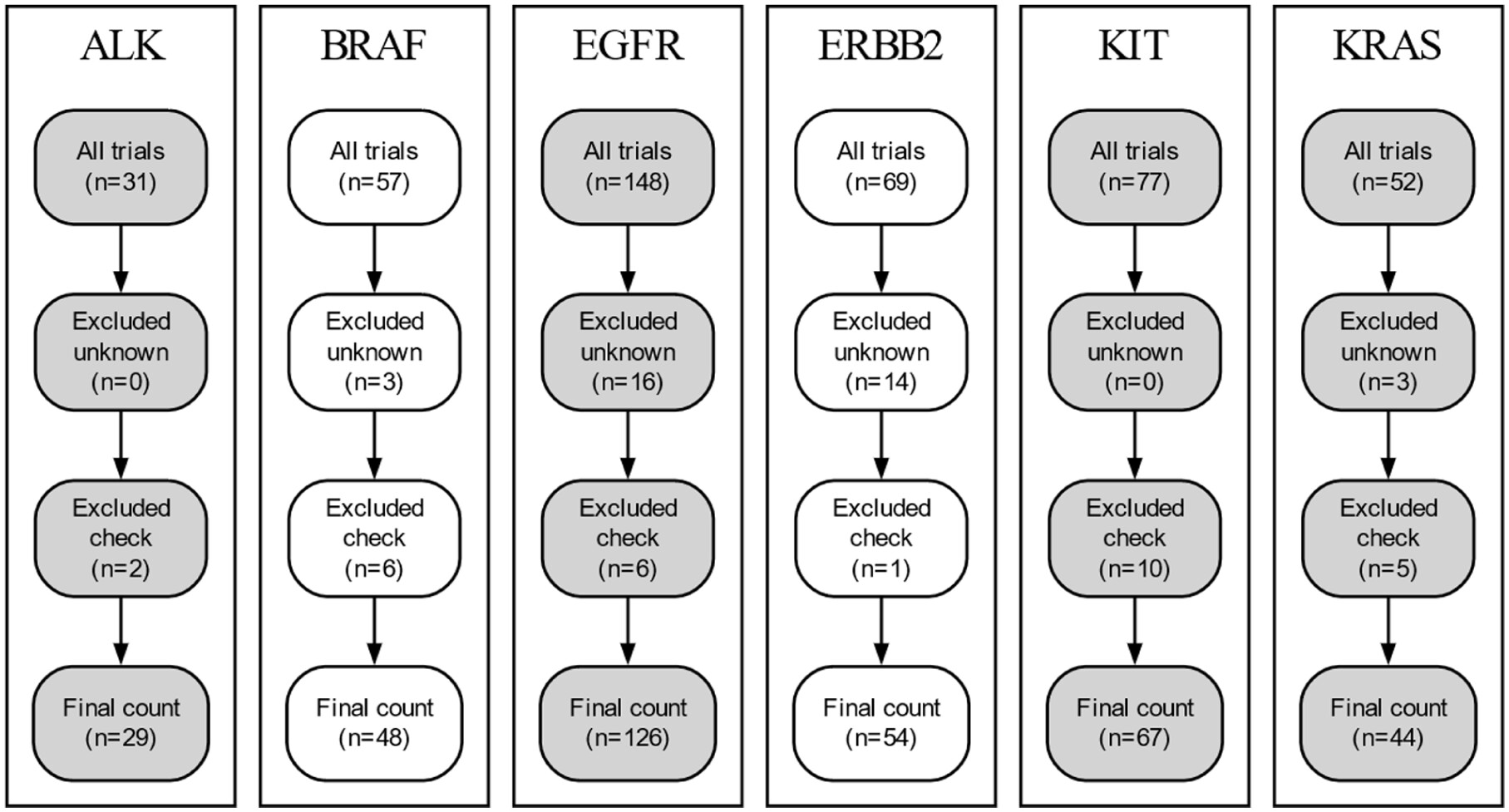
Clinical Trial Collection
| Regular expressions |
|---|
| [a-zA-Z]+[\d]+[a-zA-Z/]*[GENE MUT]+ |
| p.[a-zA-Z]+[\d]+[a-zA-Z/]*[GENE MUT]+ |
| [a-zA-Z]+[\d]+[a-zA-Z/]+[GENE MUT]+ |
| p.[a-zA-Z]+[\d]+[a-zA-Z/]+[GENE MUT]+ |
LLM Prompting and Decision Tree Exploration
| Gene | Not_wanted (no) | Wanted (yes) | Total |
|---|---|---|---|
| ALK | 16 | 13 | 29 |
| BRAF | 9 | 39 | 48 |
| EGFR | 49 | 77 | 126 |
| ERBB2 | 12 | 42 | 54 |
| KIT | 59 | 8 | 67 |
| KRAS | 17 | 27 | 44 |
| Total | 162 | 206 | 368 |


| ALK | |||
|---|---|---|---|
| Gold std/Gemini 2.0 | Precision | Recall | F1-score |
| Wanted | 0.37, std = 0.34 | 0.37, std = 0.34 | 0.37, std = 0.34 |
| Not_wanted | 0.62, std = 0.13 | 0.75, std = 0.14 | 0.67, std = 0.10 |
| Weighted avg overall | 0.51, std = 0.22 | 0.59, std = 0.14 | 0.54, std = 0.19 |
| BRAF | |||
| Gold std/GPT 4.0 | Precision | Recall | F1-score |
| Wanted | 0.80, std = 0.04 | 0.90, std = 0.11 | 0.84, std = 0.05 |
| Not_wanted | 0.00, std = 0.00 | 0.00, std = 0.00 | 0.00, std = 0.00 |
| Weighted avg overall | 0.65, std = 0.07 | 0.73, std = 0.07 | 0.68, std = 0.04 |
| EGFR | |||
| Gold std/GPT 4.0 | Precision | Recall | F1-score |
| Wanted | 0.59, std = 0.13 | 0.65, std = 0.20 | 0.61, std = 0.15 |
| Not_wanted | 0.35, std = 0.29 | 0.28, std = 0.24 | 0.30, std = 0.24 |
| Weighted avg overall | 0.49, std = 0.19 | 0.51, std = 0.18 | 0.49, std = 0.17 |
| ERBB2 | |||
| Gold std/GPT 4.0 | Precision | Recall | F1-score |
| Wanted | 0.79, std = 0.05 | 0.81, std = 0.06 | 0.79, std = 0.04 |
| Not_wanted | 0.88, std = 0.04 | 1.00, std = 0.00 | 0.94, std = 0.02 |
| Weighted avg overall | 0.67, std = 0.07 | 0.68, std = 0.06 | 0.67, std = 0.06 |
| KIT | |||
| Gold std/GPT 4.0 | Precision | Recall | F1-score |
| Wanted | 0.00, std = 0.00 | 0.00, std = 0.00 | 0.00, std = 0.00 |
| Not_wanted | 0.93, std = 0.07 | 0.87, std = 0.13 | 0.89, std = 0.07 |
| Weighted avg overall | 0.77, std = 0.07 | 0.88, std = 0.04 | 0.82, std = 0.06 |
| KRAS | |||
| Gold std/GPT 4.0 | Precision | Recall | F1-score |
| Wanted | 0.68, std = 0.09 | 0.93, std = 0.09 | 0.78, std = 0.03 |
| Not_wanted | 0.63, std = 0.41 | 0.30, std = 0.24 | 0.37, std = 0.24 |
| Weighted avg overall | 0.68, std = 0.17 | 0.68, std = 0.06 | 0.62, std = 0.11 |
| Gold std/Gemini 2.0 | Precision | Recall | F1-score |
|---|---|---|---|
| ALK | |||
| Wanted | 0.83, std = 0.24 | 0.67, std = 0.33 | 0.70, std = 0.24 |
| Not_wanted | 0.77, std = 0.23 | 0.87, std = 0.18 | 0.80, std = 0.16 |
| Weighted avg overall | 0.81, std = 0.19 | 0.76, std = 0.19 | 0.75, std = 0.20 |
| BRAF | |||
| Wanted | 0.86, std = 0.08 | 0.74, std = 0.09 | 0.79, std = 0.05 |
| Not_wanted | 0.44, std = 0.17 | 0.43, std = 0.18 | 0.43, std = 0.17 |
| Weighted avg overall | 0.75, std = 0.10 | 0.68, std = 0.09 | 0.70, std = 0.07 |
| EGFR | |||
| Wanted | 0.65, std = 0.10 | 0.66, std = 0.10 | 0.65, std = 0.10 |
| Not_wanted | 0.379, std = 0.092 | 0.411, std = 0.104 | 0.391, std = 0.088 |
| Weighted avg overall | 0.57, std = 0.13 | 0.57, std = 0.12 | 0.57, std = 0.13 |
| ERBB2 | |||
| Wanted | 0.84, std = 0. | 0.76, std = 0.09 | 0.79, std = 0.07 |
| Not_wanted | 0.31, std = 0.19 | 0.47, std = 0.36 | 0.36, std = 0.22 |
| Weighted avg overall | 0.72, std = 0.16 | 0.69, std = 0.10 | 0.69, std = 0.12 |
| KIT | |||
| Wanted | 0.37, std = 0.41 | 0.50, std = 0.50 | 0.37, std = 0.34 |
| Not_wanted | 0.93, std = 0.07 | 0.87, std = 0.13 | 0.89, std = 0.07 |
| Weighted avg overall | 0.87, std = 0.10 | 0.82, std = 0.11 | 0.83, std = 0.10 |
| KRAS | |||
| Wanted | 0.70, std = 0.10 | 0.93, std = 0.10 | 0.79, std = 0.08 |
| Not_wanted | 0.63, std = 0.41 | 0.35, std = 0.25 | 0.43, std = 0.29 |
| Weighted avg overall | 0.68, std = 0.19 | 0.70, std = 0.11 | 0.65, std = 0.15 |
| Gene mutation | GPT4 F1-score | Gemini 2.0 F1-score | P-value |
|---|---|---|---|
| ALK | 0.75, std = 0.20 | 0.54, std = 0.19 | P < .001 |
| BRAF | 0.68, std = 0.04 | 0.70, std = 0.07 | No statistical difference |
| EGFR | 0.49, std = 0.17 | 0.57, std = 0.13 | P < .001 |
| ERBB2 | 0.67, std = 0.06 | 0.69, std = 0.12 | No statistical difference |
| KIT | 0.82, std = 0.06 | 0.83, std = 0.10 | No statistical difference |
| KRAS | 0.62, std = 0.11 | 0.65, std = 0.15 | No statistical difference |
| Gene name | Criterion | Maximum depth | Minimum samples per leaf | Minimum samples split |
|---|---|---|---|---|
| ALK | Gini | 10 | 2 | 10 |
| BRAF | Gini | None | 1 | 2 |
| EGFR | Gini | 10 | 1 | 2 |
| ERBB2 | Entropy | 40 | 2 | 2 |
| KIT | Entropy | 20 | 2 | 2 |
| KRAS | Gini | 40 | 4 | 10 |
| Gold std/GPT 4.0 | Precision | Recall | F1-score |
|---|---|---|---|
| ALK | |||
| Yes | 0.94 | 1.0 | 0.97 |
| No | 1.0 | 0.96 | 0.96 |
| Weighted avg overall | 0.97 | 0.97 | 0.97 |
| BRAF | |||
| Yes | 1.0 | 1.0 | 1.0 |
| No | 1.0 | 1.0 | 1.0 |
| Weighted avg overall | 1.0 | 1.0 | 1.0 |
| EGFR | |||
| Yes | 1.0 | 1.0 | 1.0 |
| No | 1.0 | 1.0 | 1.0 |
| Weighted avg overall | 1.0 | 1.0 | 1.0 |
| ERBB2 | |||
| Yes | 0.86 | 1.00 | 0.92 |
| No | 1.0 | 0.95 | 0.98 |
| Weighted avg overall | 0.97 | 0.96 | 0.96 |
| KIT | |||
| Yes | 0.98 | 1.00 | 0.99 |
| No | 1.0 | 0.88 | 0.93 |
| Weighted avg overall | 0.99 | 0.99 | 0.98 |
| KRAS | |||
| Yes | 0.77 | 1.00 | 0.87 |
| No | 1.0 | 0.81 | 0.90 |
| Weighted Avg Overall | 0.89 | 0.91 | 0.88 |
| ALK | |||
|---|---|---|---|
| Gold std/Gemini | Precision | Recall | F1-score |
| Yes | 1.0 | 1.0 | 1.0 |
| No | 1.0 | 1.0 | 1.0 |
| Weighted avg overall | 1.0 | 1.0 | 1.0 |
| BRAF | |||
| Gold std/GPT 4.0 | Precision | Recall | F1-score |
| Yes | 0.89 | 0.89 | 0.89 |
| No | 0.97 | 0.97 | 0.97 |
| Weighted avg overall | 0.96 | 0.96 | 0.96 |
| EGFR | |||
| Gold std/GPT 4.0 | Precision | Recall | F1-score |
| Yes | 0.96 | 1 | 0.98 |
| No | 1.0 | 0.97 | 0.99 |
| Weighted avg overall | 0.98 | 0.98 | 0.98 |
| ERBB2 | |||
| Gold std/GPT 4.0 | Precision | Recall | F1-score |
| Yes | 1.0 | 1.0 | 1.0 |
| No | 1.0 | 1.0 | 1.0 |
| Weighted avg overall | 1.0 | 1.0 | 1.0 |
| KIT | |||
| Gold std/GPT 4.0 | Precision | Recall | F1-score |
| Yes | 1.0 | 1.0 | 1.0 |
| No | 1.0 | 1.0 | 1.0 |
| Weighted avg overall | 1.0 | 1.0 | 1.0 |
| KRAS | |||
| Gold std/GPT 4.0 | Precision | Recall | F1-score |
| Yes | 1.0 | 1.0 | 1.0 |
| No | 1.0 | 1.0 | 1.0 |
| Weighted avg overall | 1.0 | 1.0 | 1.0 |
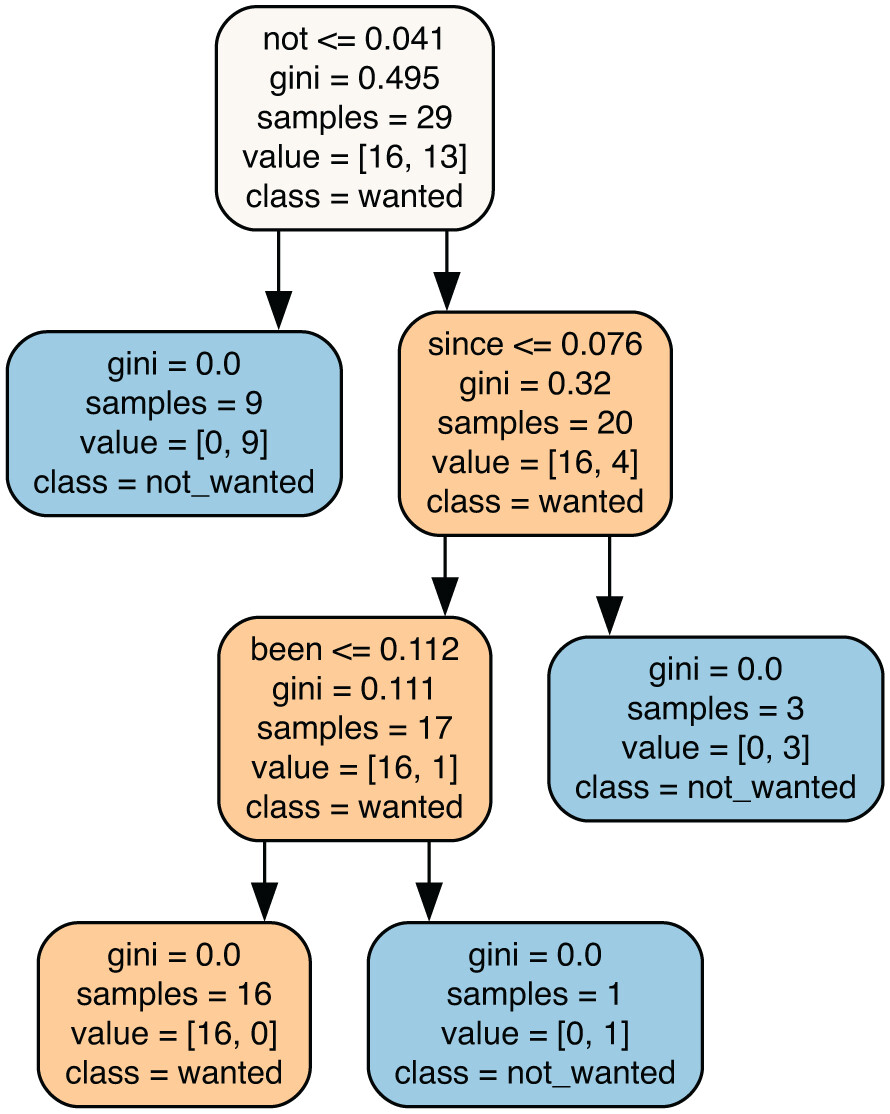
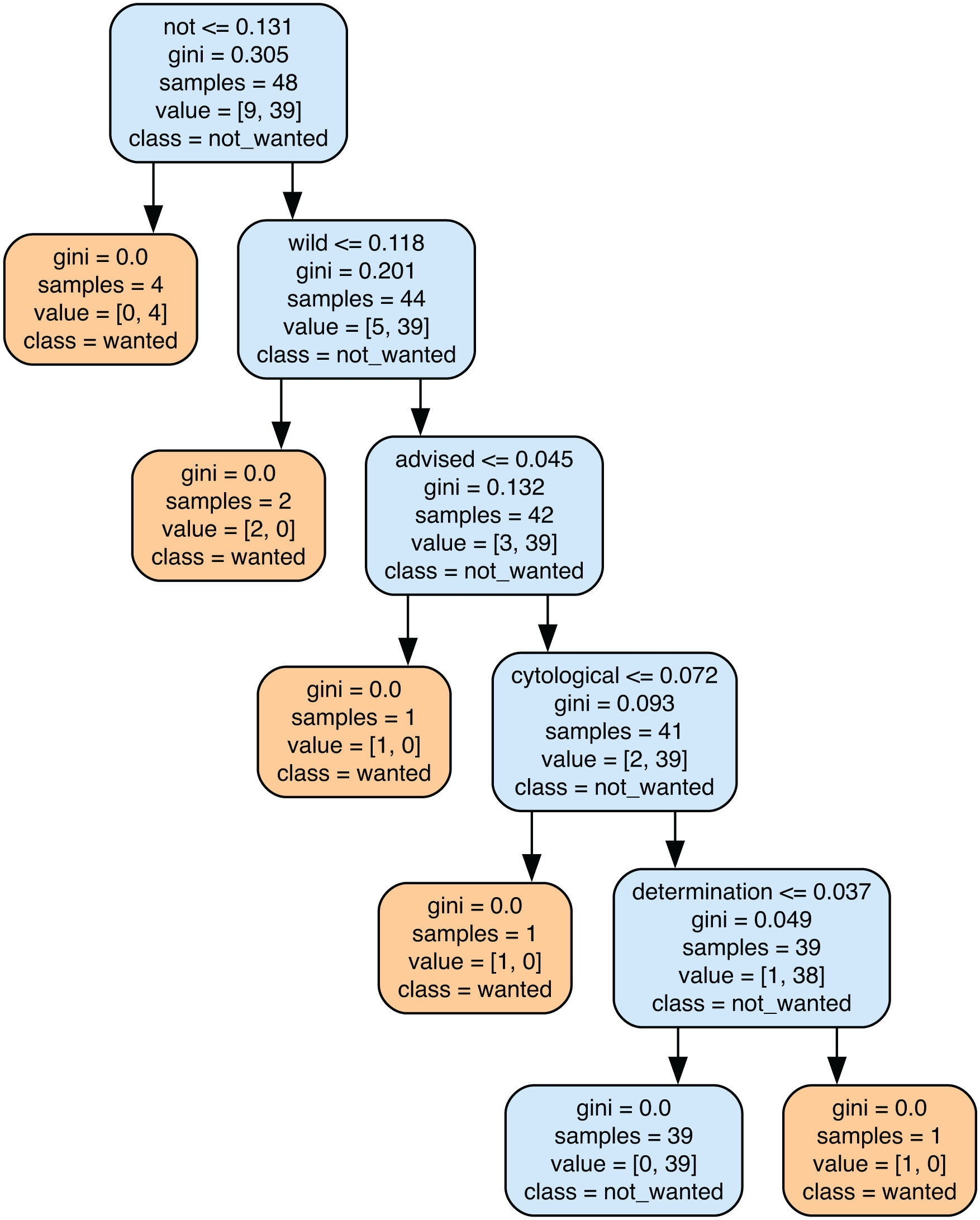

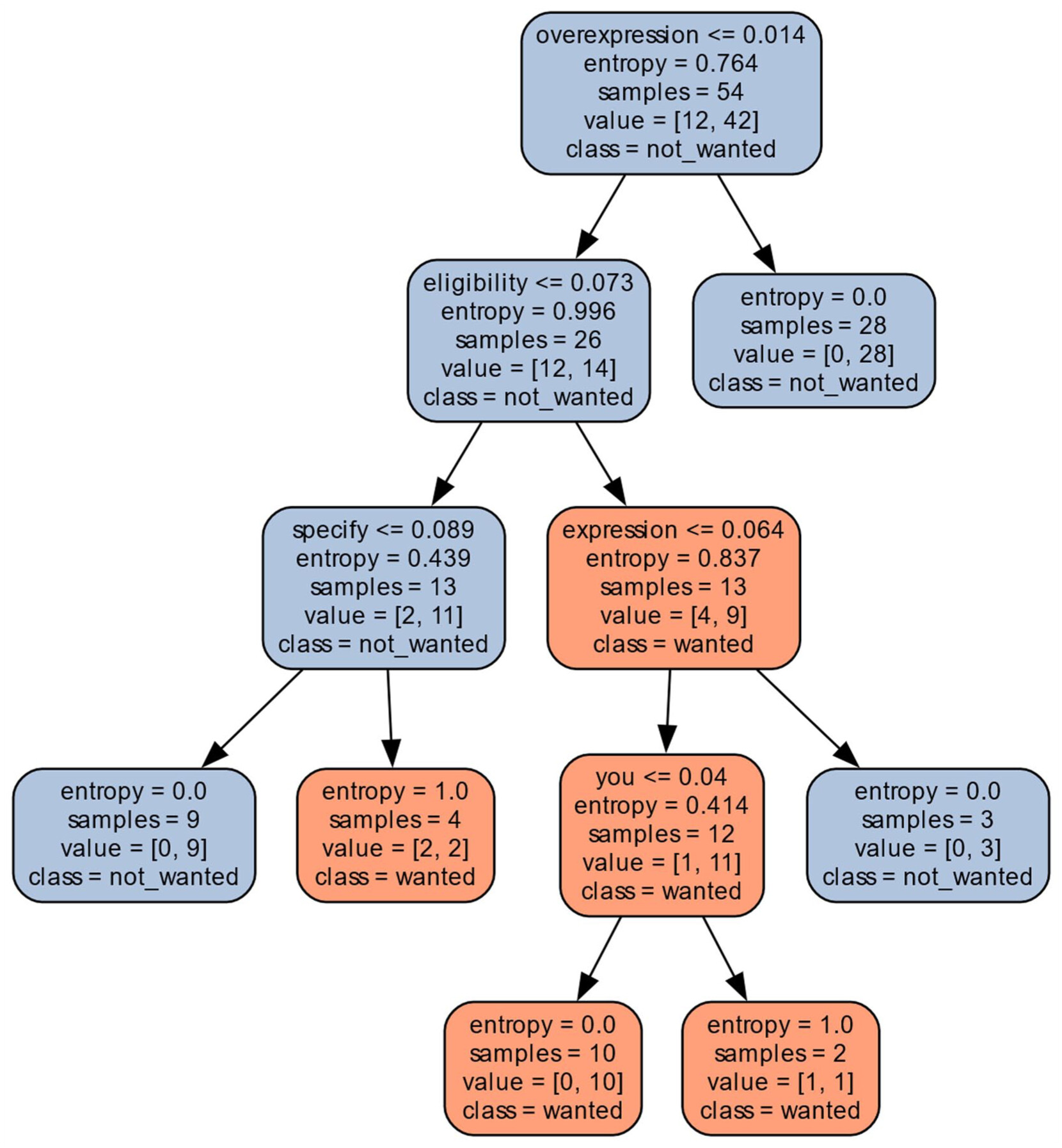

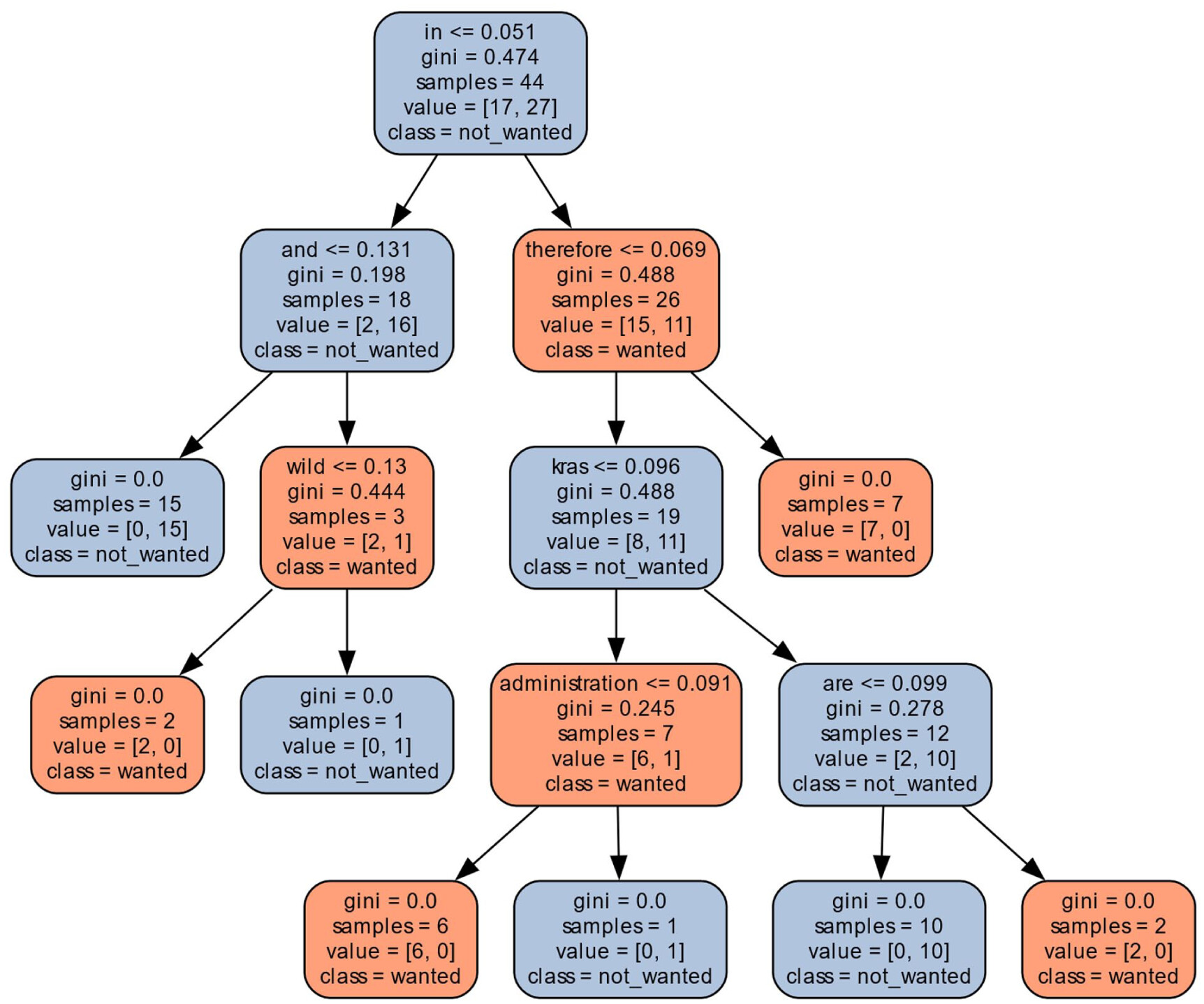
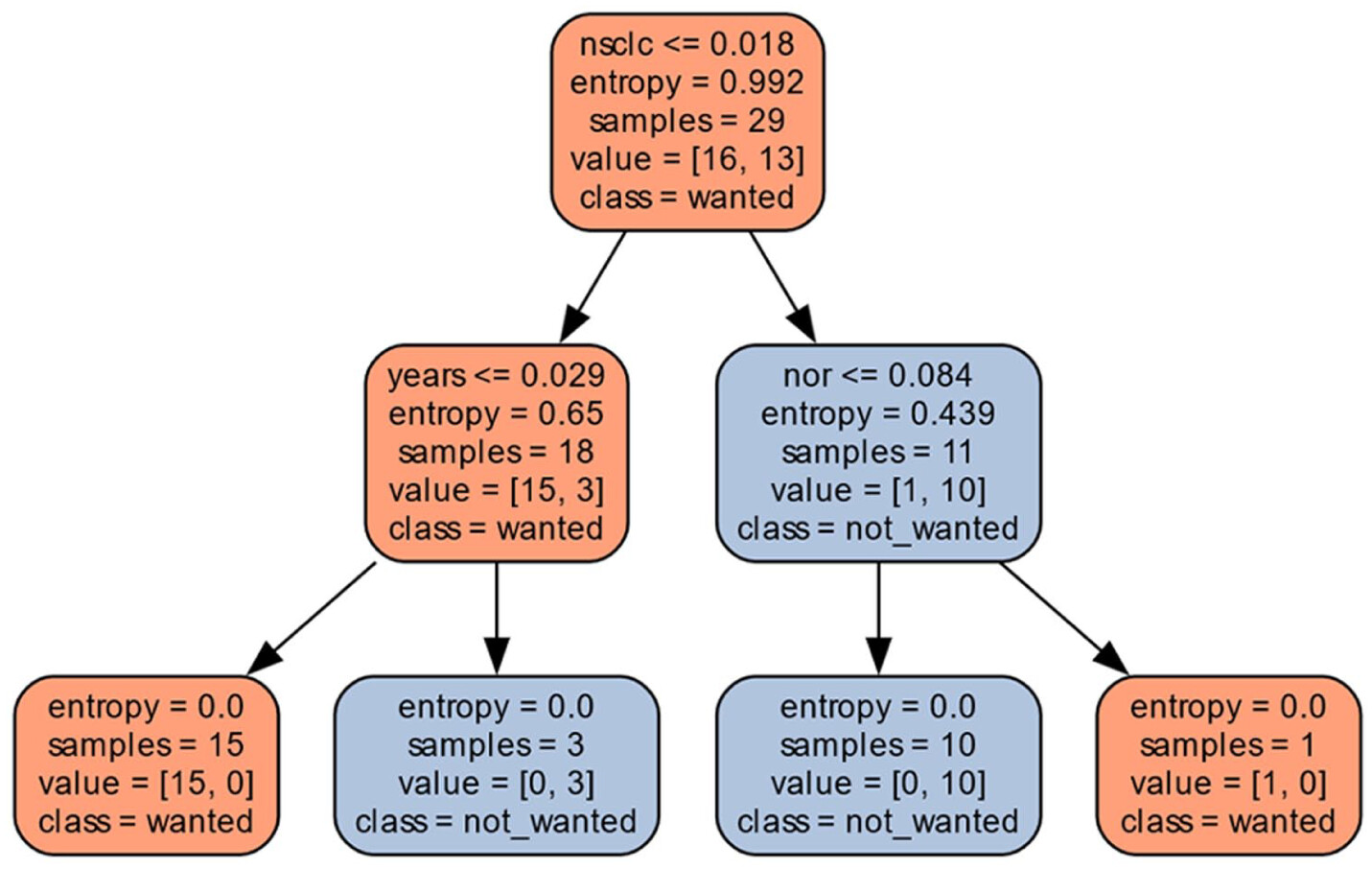
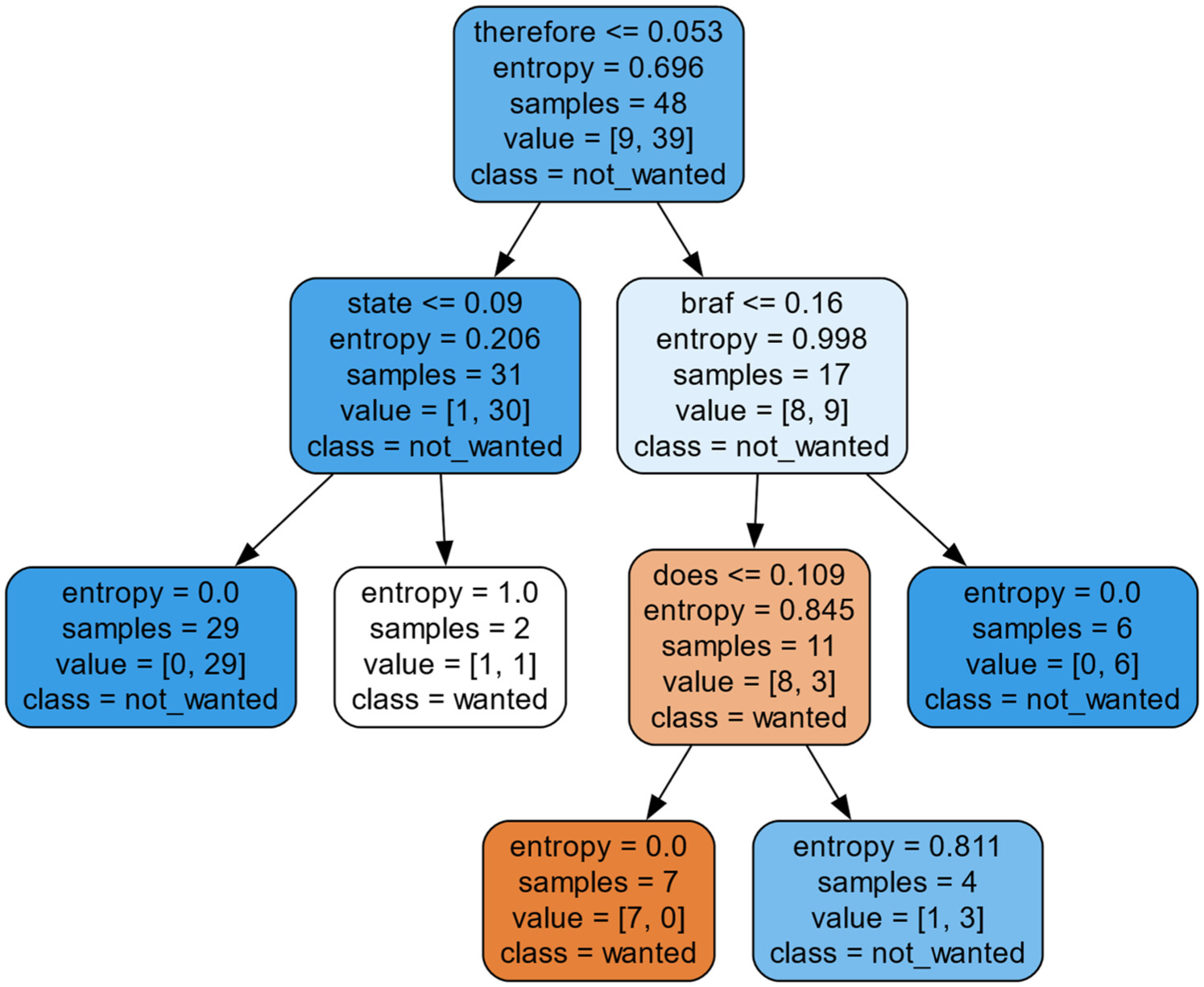
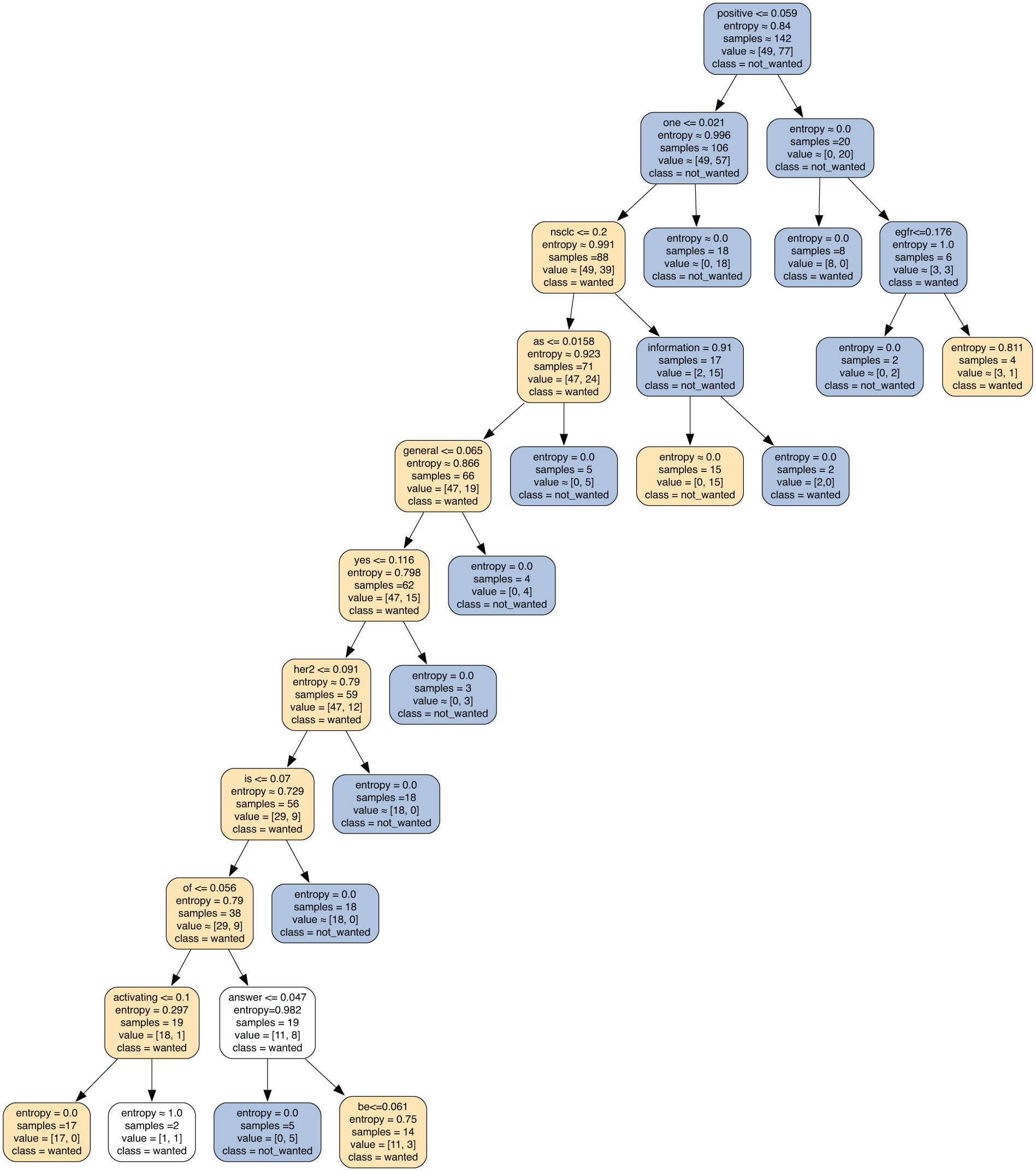

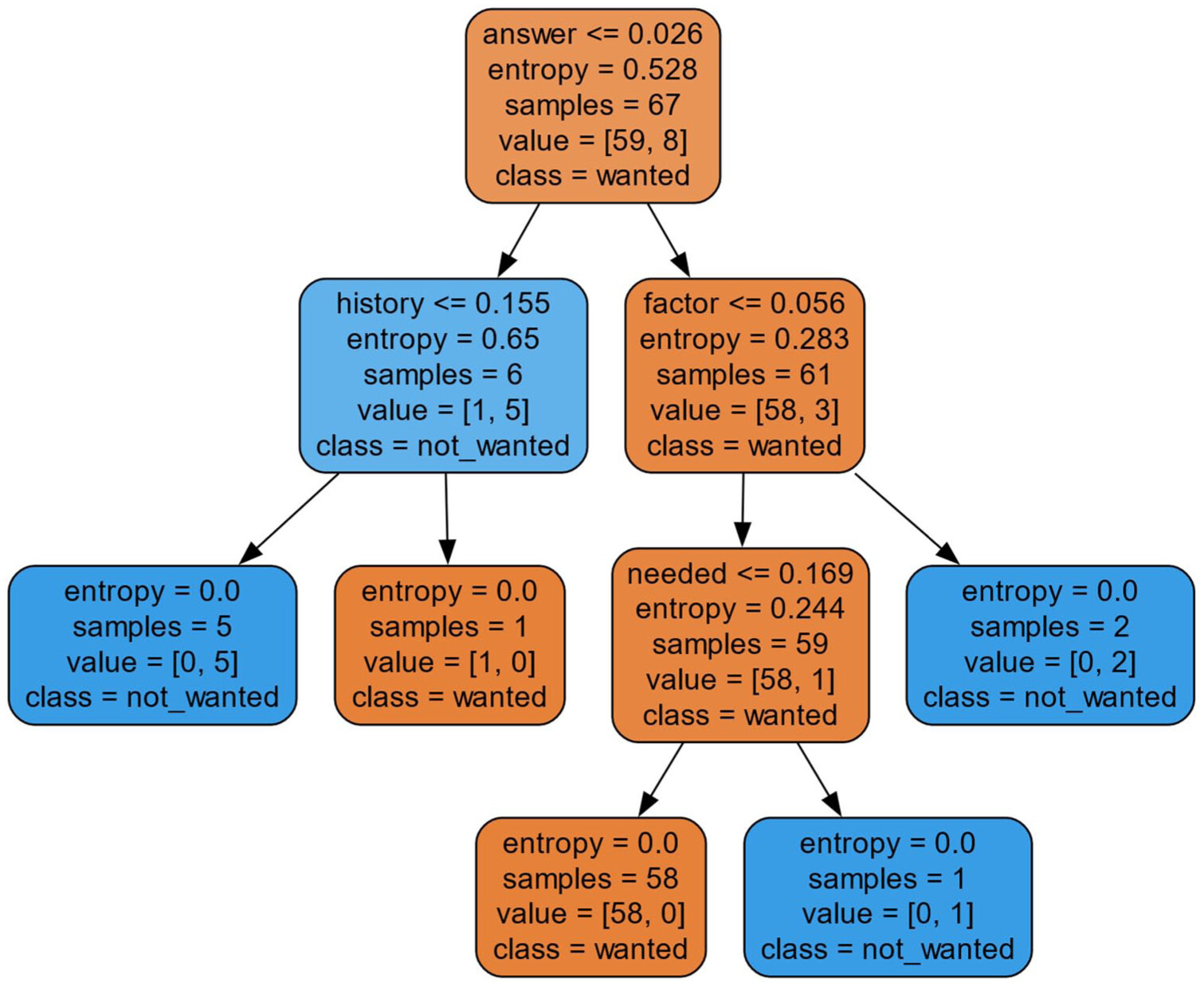
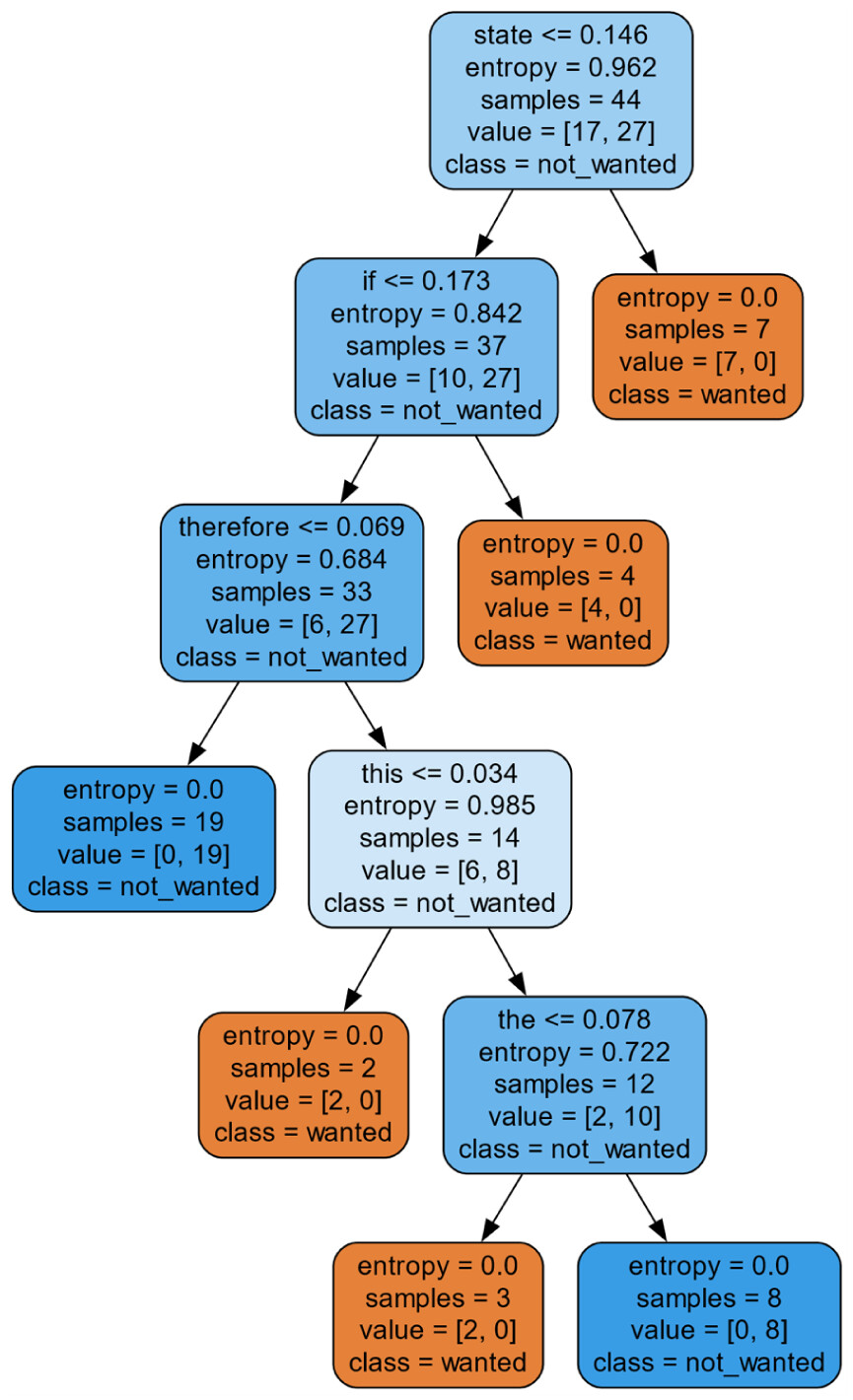
Results
| Gene name | Criterion | Maximum depth | Minimum samples per leaf | Minimum samples split |
|---|---|---|---|---|
| ALK | Entropy | 40 | 1 | 10 |
| BRAF | Entropy | 20 | 2 | 10 |
| EGFR | Entropy | 20 | 2 | 5 |
| ERBB2 | Gini | 30 | 1 | 2 |
| KIT | Entropy | None | 1 | 2 |
| KRAS |
"eligibilityCriteria": "* Histologically or cytologically diagnosed NSCLC, all histologies are acceptable.* Patients can be included in the study with any disease stage and at any time during the disease course.* Any type (surgery, RadioTherapy, chemotherapy, targeted agents) of previous treatment and any line of treatment are eligible. . .
"eligibilityCriteria": "Inclusion Criteria:* Histologically confirmed colorectal adenocarcinoma;* Availability of formalin-fixed paraffin embedded tumor block from primary or metastasis;* KRAS and BRAF wild-type status of primary colorectal cancer or related metastasis;* :Question:Based on the following clinical trial information, would a patient with a BRAF gene mutation be eligible for the clinical trial?Answer:Based on the eligibility criteria provided for the clinical trial, a patient with a BRAF gene mutation would not be eligible to participate. The inclusion criteria specifically require "KRAS and BRAF wild-type status of primary colorectal cancer or related metastasis." This means the patient’s colorectal cancer must not have mutations in either the KRAS or BRAF genes to be eligible for the trial. Therefore, a patient with a BRAF gene mutation would not meet these criteria and would be considered ineligible for treatment.
Question:Based on the following clinical trial information, would a patient with a BRAF gene mutation be eligible for the clinical trial?Answer:No. While the criteria state that patients with NSCLC (Non-Small Cell Lung Cancer) at any stage and with any prior treatment are eligible, the *studyPopulation* section specifies that this trial is for patients with proven EGFR mutations or those with wild-type EGFR (no EGFR mutations). The information provided describes a study looking at EGFR, not BRAF. Therefore, a patient with a BRAF mutation would *not* be eligible for this particular trial.
Question:Based on the following clinical trial information, would a patient with an ERBB2 gene mutation be eligible for the clinical trial?Answer:Based on the clinical trial information provided, the eligibility for the trial is primarily based on HER2 status and prior treatment history. The trial mentions inclusion criteria for HER2-positive breast cancer patients, which are determined by either IHC3+ or FISH+ assays, indicating overexpression or amplification of the HER2 protein.The ERBB2 gene, also known as HER2, is responsible for the production of the HER2 protein. Therefore, if a patient has an ERBB2 gene mutation that results in HER2 positivity (such as overexpression or amplification), they would meet the HER2-positive criteria part of the inclusion.
"eligibilityCriteria": "Inclusion Criteria:* Concurrent or prior malignancy, other than curatively treated carcinoma-in-situ or basal cell carcinoma of the skinQuestion:Based on the following clinical trial information, would a patient with a ERBB2 gene mutation be eligible for the clinical trial?Answer:This trial is for patients with neurofibromatosis type 2 (NF2), indicated by the inclusion criteria requiring a diagnosis of NF2. The criteria mention nothing about ERBB2 mutations. . . . To be eligible, they would need to meet *all* other inclusion criteria and *none* of the exclusion criteria.
“eligibilityCriteria”: “Exclusion Criteria” . . . Co-morbid systemic illnesses or other severe concurrent disease which, in the judgment of the investigator, would make the patient inappropriate for entry into this study or interfere significantly with the proper assessment of safety and toxicity of the prescribed regimensQuestion:Based on the following clinical trial information, would a patient with a KIT gene mutation be eligible for the clinical trial?Answer:Based on the provided eligibility criteria for the clinical trial, a patient with a KIT gene mutation would not automatically be excluded from participation. The inclusion criteria specifically mention acute myeloid leukemia (AML) with Kit (CD117) expression, which can be a common feature in patients with a KIT gene mutation. The trial focuses on KIT expression rather than directly on the presence or absence of a KIT gene mutation.However, it is important to note that there are multiple inclusion and exclusion criteria that must be met or avoided. The patient’s overall health status, previous treatments, and concurrent medications, among other factors, must comply with the trial’s requirements as listed in the other criteria.
Exclusion criteria: . . . Male patients with a partner of childbearing potential must agree to use contraception in addition to having their partner use another contraceptive method during the trial. . . .Any of the following in 3 months before inclusion: grade 3-4 gastrointestinal bleeding (unless due to resected tumor), treatment resistant peptic ulcer disease, erosive esophagitis or gastritis, infectious or inflammatory bowel disease, or diverticulitis* Other concomitant or previous malignancy, except: * adequately treated in-situ carcinoma of the uterine cervix * basal or squamous cell carcinoma of the skin * cancer in complete remission for \\>5 years*. . .Question:Based on the following clinical trial information, would a patient with a KRAS gene mutation be eligible for the clinical trial?Answer:Based on the eligibility criteria provided for the clinical trial, there is no specific mention of a KRAS gene mutation being a criterion for exclusion or inclusion. The trial does not specify genetic markers like KRAS mutations as part of the eligibility or exclusion criteria. Therefore, a patient with a KRAS gene mutation would still be eligible as long as they meet the other inclusion criteria and do not meet any of the exclusion criteria listed
Discussion
Limitations and Future Work
Conclusion
Ethical Considerations
Consent to Participate
Declaration of Conflicting Interests
Funding
ORCID iD
References
Cite
Cite
Cite
Download to reference manager
If you have citation software installed, you can download citation data to the citation manager of your choice
Information, rights and permissions
Information
Published In

Keywords
Rights and permissions
Authors
Author Contributions
Metrics and citations
Metrics
Publication usage*
Total views and downloads: 202
*Publication usage tracking started in December 2016
Publications citing this one
Receive email alerts when this publication is cited
Web of Science: 0
Crossref:
There are no citing articles to show.
Figures and tables
Figures & Media
Tables
View Options
View options
PDF/EPUB
View PDF/EPUBAccess options
If you have access to journal content via a personal subscription, university, library, employer or society, select from the options below:
I am signed in as:
View my profileSign out

I can access personal subscriptions, purchases, paired institutional access and free tools such as favourite journals, email alerts and saved searches.
loading institutional access options
Alternatively, view purchase options below:
Access journal content via a DeepDyve subscription or find out more about this option.
