Introduction
Heterocyclic compounds are the cyclic organic compounds which contain at least one heteroatom; the most common heteroatoms are the nitrogen, oxygen and sulfur.
1 Nitrogen-containing heterocyclic compounds and their derivatives have historically been invaluable as a source of therapeutic agents.
2 A variety of bis-heterocyclic compounds comprising N, S and O have been studied in industry as liquid crystals and in medicinal chemistry as biologically active compounds with properties, such as antifungal, anti-inflammatory, antibacterial, antioxidant and anticancer activity.
3,4 New anticancer medication has been significantly influenced by bis-heterocyclic compounds.
5,6Pyrazole, which has two nitrogen atoms and aromatic character, provides diverse functionality and stereochemical complexity in a five-membered ring.
7 Pyrazole derivatives containing phenyl group remarkably display a variety of biological activities. Pyrazolines can be prepared by reactions of hydrazine hydrate or diazoalkanes with α,β-unsaturated carbonyl compounds.
8,9Curcumin is a widely studied biologically active, bis-phenolic, enolic diketone. Modification of the diketone functionality has been investigated, and numerous monocarbonyl curcumin analogues with increased stability and activity were prepared including a variety of 1, 3-diphenylprop-2-en-1-ones.
10–12The biological activities of these synthetic curcumin analogues are mediated by their distinct unsaturated carbonyl functionality. They have been licenced for use in medicine to treat a variety of conditions, including the choleretic metochalcone and the sofalcone antiseptic mucoprotective ulcer.
12 However, bis-chalcones derivatives potentially have more physical and biological applications because of the larger number of their derivatives. As a result, bis-chalcones and their derivatives have recently been widely studied for their biological activities.
13There are several ways to prepare bis-chalcones, including the use of microwaves. Microwave heating can speed up reactions to a few minutes rather than hours or days.
14 Several studies have reported the synthesis of bis-pyrazolines and bis-pyrimidines from bis-chalcones using microwave heating and ultrasound irradiation.
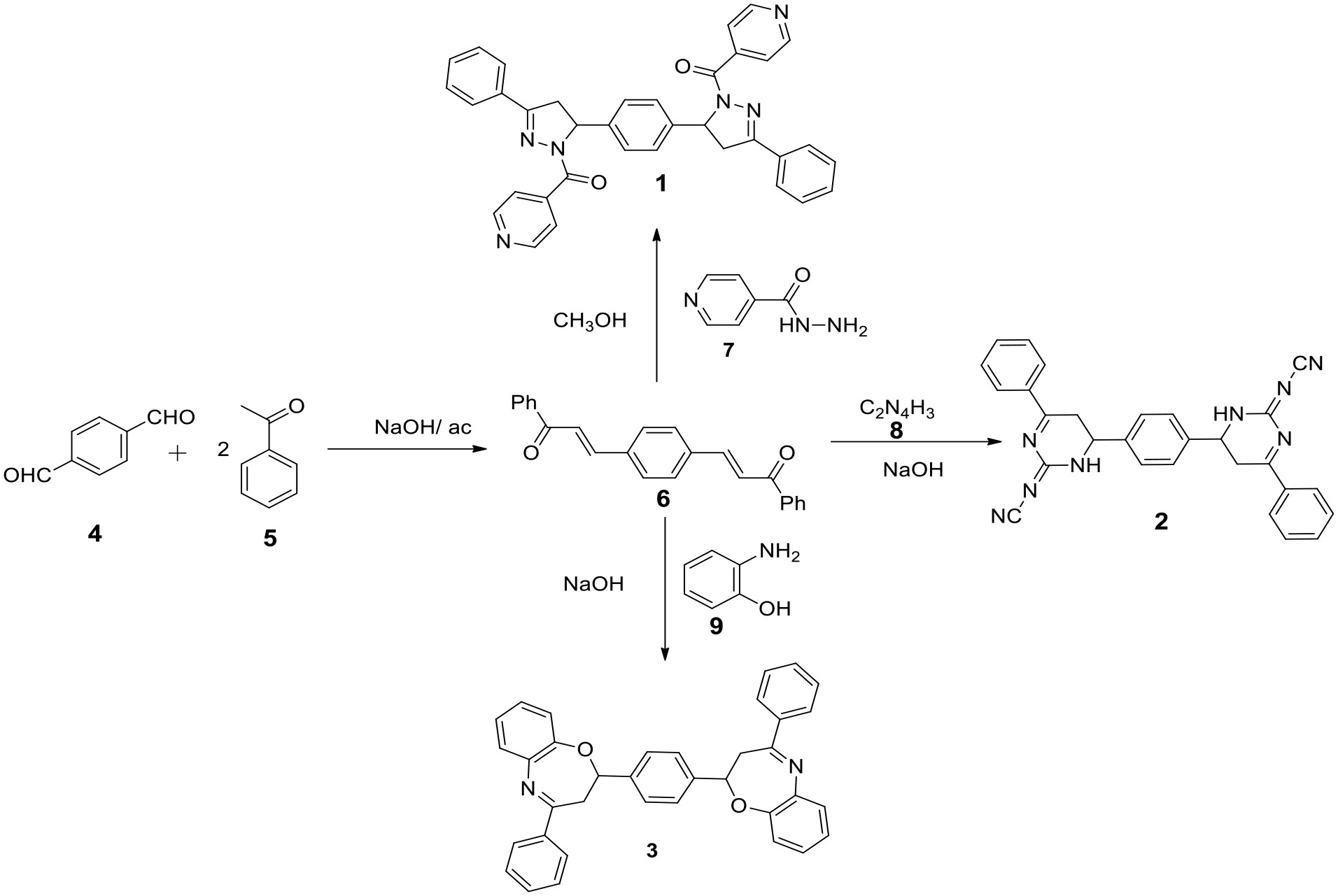
15–17We here report the design and synthesis of three new bis-heterocyclic compounds from a bis-chalcone that was prepared by the reaction between acetophenone and terephthalaldehyde using the Claisen–Schmidt method. The title compounds were tested and their antimicrobial activities on three bacteria and two types of fungi were studied.
Results and discussion
The new bis-heterocyclic compounds containing pyrazoline, pyrimidine, and oxazepine heterocycles were synthesized from the bis-chalcone. Acetophenone was used to prepare the bis-chalcone. Assembly of the heterocycles was carried out by bis-Michael addition on the bis-chalcone followed by dehydration to obtain the target bis-heterocyclic compounds (
Scheme 1). The reactions were monitored by TLC. The structures of the compounds prepared were confirmed by spectroscopic methods, such as
1H NMR,
13C NMR and Fourier transform infrared (FTIR). The biological effectiveness of the products on two types of bacteria was studied. They showed good biological activities towards gram-positive and gram-negative bacteria.
The bis-chalcone
6 was synthesized by bis-aldol condensation following the literature procedure and was isolated as a bright yellow solid; 89%; m.p. 195 °C–196 °C.
18,19 Reactions of the bis-chalcone
6 with Reagents
7–
9 gave the target Compounds
1–
3 in good yields.
The structure of Compound
1 was confirmed by the presence of the amide carbonyl stretching vibration at 1660 cm
−1 in its infrared (IR) spectrum. The absorption band around 1415 cm
−1 was assigned to the C=N stretching vibration. The absence of carbonyl band clearly supported the formation of the target compound. In addition, characteristic absorption frequencies around 3057–3030 cm
−1 were assigned to aromatic CH stretching vibrations.
In its
1H NMR spectrum, the aromatic protons were observed as multiplets at δ 7.06–8.80 ppm. The pyrazoline showed a 4H multiplet at δ 3.70 ppm, and a 2H multiplet at δ 4.8 ppm rather than a distinct double-doublet because of the presence of two diastereoisomers due to the remote stereogenic centres.
20,21In the 13C NMR spectrum, the C4 carbon of the pyrazoline appeared at δ 65 ppm. The C3 carbon of the pyrazoline observed at δ 45 ppm. The aromatic carbons were observed in the region of 122–140 ppm. In addition, the signal belonging to the carbonyl group was observed at δ 164.3 ppm.
The structure of Compound 2 was consistent with the disappearance of the carbonyl group and appearance of the absorption 1445 cm−1 that was assigned to the C=N bond in its IR spectrum. The absorption at 2250 cm−1 was attributed to the C=N stretching vibration. In its 1H NMR spectrum, the doublet at δ 2.4 ppm was assigned to the NH of the dihydropyrimidines, the aromatic protons were observed as a multiplet at δ 7.25–7.75 ppm and those of the dihydropyrimidine at δ 4.8 and 3.7 ppm. In its 13C NMR spectrum, the peaks at δ 55 and 45 ppm were attributed to the dihydropyrimidines, those at δ 153 and 163 ppm assigned to the C=N groups, the aromatic carbons were observed at the range δ 115–143 ppm, and the cyanide carbon at δ 112 ppm.
The formation of Compound 3 was consistent with the IR peak of the dihydrobenzooxazepine C=N at 1442 cm−1 with peaks around 2900–2800 cm−1 for the aromatic C–H. In its 1H NMR spectrum, the aromatic protons were at δ 6.40–7.60 ppm with the methylene and methine protons at δ 3.01 and 5.45 ppm, respectively. In its 13C NMR spectrum, peak at δ 172.67 ppm was assigned to the C=N carbon, while the signals due to the aromatic carbons were in the range δ 115–145 ppm. The carbons of the CH and CH2 of the dihydrobenzooxazepine appeared at δ 76 and 48 ppm, respectively.
Biological activity
The biological activity of the compounds prepared was investigated by their effect on three bacteria
Escherichia coli,
Klebsiella pneumonia and
Staphylococcus aureus by diffusion in an agar medium and nutrient.
22 The compounds prepared showed a significant, clear and selective effect against the
Staphylococci when compared to the results found for the other bacteria.
23From these results, Compound
1 showed an inhibitory effect with its best effect at a concentration of 150 mg/mL. Compound
2 exhibited acceptable inhibition of the three bacteria at all concentrations and Compound
3 showed the best biological activity of the three compounds on all three bacteria, possibly due to the dihydrooxazepine which interferes with the metabolic activity in the cytoplasm of bacteria cells (
Figure 1).
Three types of bacteria were used, two of which were gram-negative (E. coli, K. pneumonia) and another gram-positive, S. aureus. The culture medium used was Mueller–Hinton Agar (MHA), which is used in the determination of the biological activities of antibiotics and chemicals for medicinal use during measurements and determinations of minimum inhibitory concentrations (MIC). Solutions with concentrations of 50, 100 and 150 mg/mL were prepared using dimethyl sulfoxide (DMSO) as the solvent for the compounds being investigated. The diffusion method was used in the MHA media.
The study was carried out by following the Agar well diffusion method after inoculating the culture medium with bacterial isolates. Drilling was done in the dishes using the metric method
24,25 cylinder according to the US Pharmacopoeia 35 USP, with the help of a borer cork. Then, 40 µL of the solutions prepared for the three concentrations were placed in each of the drill bits. They were incubated in the incubator at a temperature of 37 °C for 24 h, then the results were read after 24 h to show the effect of the three compounds on the diameter of the apparent inhibition in the dishes around the different holes. The increase in the diameter of the inhibition means an increase in the biological activity of the different compounds and were compared that with the inhibition diameters of the standard antibiotics. Tamm led the use of the antibiotic isoniazid as a control sample, based on what is used in the Ministry of Health laboratories and the World Health Organization examinations.
Experiment
Material and methods
The following reagents and solvents were purchased from Merck without further purification: 2-aminophenol, isoniazid, cyanoguanidine, acetophenone, terephthalaldehyde, methanol, sodium hydroxide, tetrahydrofuran, and dioxane. Merck Silica Gel 60 F254 was used for column chromatography, whereas Merck Silica Gel 60 (0.063–0.200 mm) was used for thin-layer chromatography on aluminium plates (20 × 20 cm2). The compounds were identified using FTIR (Shimadzu Prestige-21, KBr discs), 1H NMR (500 MHz) and 13C NMR (125 MHz) (CDCl3, standard internal TMS) spectrometers. The results were analysed statistically, using ANOVA, and comparisons of the arithmetic means of the compounds with Duncan’s multinomial test, with a probability level of p ⩽ 0.05.
(2E,2′E)-3,3′-(1,4-phenylene)bis(1-phenylprop-2-en-1-one) (6)
Acetophenone (
5) (0.04 mol) was dissolved in 1 mL of 10% sodium hydroxide with continuous stirring. Terephthalaldehyde
4 (0.02 mol) was dissolved in 10 mL of ethanol and added to the reaction mixture. This was stirred for 3 h at 20 °C–30 °C and stored for 12 h in the refrigerator. The precipitate was filtered and washed with ethanol to give Compound
6 as a bright yellow solid; 89%; m.p. 195 °C–196 °C.
18(5,5′-(1,4-phenylene)bis(3-phenyl-4,5-dihydro-1H-pyrazole-5,1-diyl))bis(pyridin-4-yl methanone) (1)
In a round flask, 0.004 mol of bis-chalcone 6 was dissolved in 10 mL of 95% ethanol. Then, 0.008 mol of pyridine-4-carboxylic hydrazide (isoniazid) (7) was added to the mixture and the mixture stirred for 3 h at 30 °C–40 °C. The mixture was then cooled, washed and recrystallized from ethanol to obtain the product as a pale orange solid; 78%; m.p. 255 °C–256 °C. 1H NMR (400 MHz, CDCl3): δ 8.8 (m, 4H, pyridine), 7.8 (m, 4H, pyridine), 7.5–7.2 (m, 10H, ArH), 4.8 (m, 2H, CH) and 3.70 (m, 4H, CH2). 13C NMR (126 MHz, CDCl3): δ 164, 157, 152, 150–125 (18C), 125, 121, 65, 45 ppm. FTIR (KBr): 3034 (C–H, aromatic), 1660 (C=O, amide), 1415 (C=N), 1521 (C=C), 1271 (C–N).
N,N′-(6,6′-(1,4-phenylene)bis(4-phenyl-5,6-dihydropyrimidin-6(1H)-yl-2(1H)-ylidene))dicyanamide (2)
Cyanoguanidine (8) (0.006 mol) was dissolved in 30 mL of ethanol and 10 mL of 10% sodium hydroxide solution was added to the solution. The mixture was stirred in an ice bath for 1 h, and then 0.003 mol of bis-chalcone (6) was added gradually. The mixture was continuously stirred for 4 h in an ice bath to complete the reaction and then the solution was left for 24 h in the refrigerator for the purpose of sedimentation. The precipitate was then removed by filtration and was recrystallized from ethanol to obtain the product as a pale yellow solid; 85%; m.p. 229 °C–228 °C. 1H NMR (400 MHz, CDCl3): δ 7.75–7.25 (m, 14H, ArH), 4.8 (m, 2H, CH), 3.7 (m, 4H, CH2) and 2.4 (m, 2H, NH). 13C NMR (126 MHz, CDCl3): δ 163, 153, 143–115 (18C), 112, 57, 45 ppm. FTIR (KBr): 3260 (C–H), 2250 (CN), 1445 (C=N), 1114 (C–N) cm−1.
1,4-bis(4-phenyl-2,3-dihydrobenzo[b][1,4]oxazepin-2-yl)benzene (3)
2-Aminophenol (9) (0.004 mol) was dissolved in 25 mL of ethanol, and 15 mL of 10% sodium hydroxide solution was added with continuous stirring for 10 min. Then the mixture was stirred in an ice bath and 0.002 mol of the bis-chalcone (6) was added gradually. The mixture was stirred for a further 4 h in an ice bath to complete the reaction and then the solution was left for 24 h in the refrigerator for the purpose of sedimentation to give the product as a solid; 81%; m.p. 224 °C–225 °C. 1H NMR (400 MHz, CDCl3): δ 7.6–6.4 (m, 22H, ArH), 5.45 (m, 2H, CH), 3.01 (m, 4H, CH2). 13C NMR (126 MHz, CDCl3): δ 172.6, 149, 145–115 (26C), 76, 48. FTIR (KBr): 2900-2800 (C–H, ArH), 1569 (C=C), 1442 (C=N), 1040 (C–O–C).