Case presentation
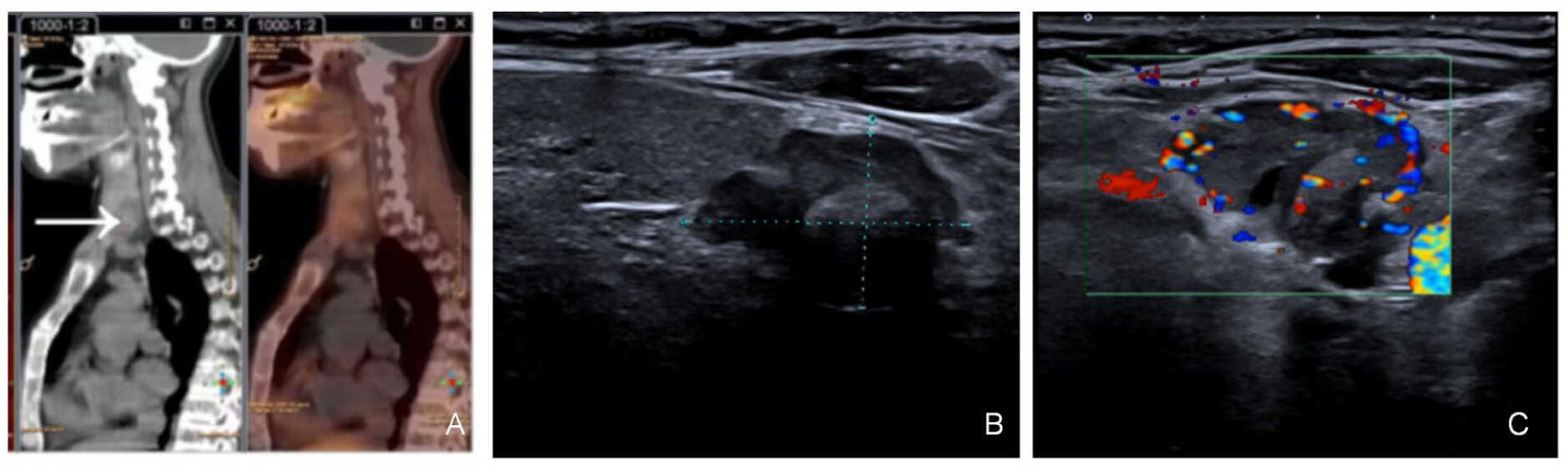
The patient is a 44-year-old woman who presented with a 2-month history of intermittent chest pain without obvious triggers. The pain progressively worsened, predominantly localized to bilateral costal arches, exacerbated by physical activity, and nonradiating. Associated symptoms included fatigue, bradykinesia (slowness of movement), and intermittent claudication during ambulation. Bone density lumbar spine examination suggested osteoporosis. She was admitted to the Department of Endocrinology of our hospital with parathyroid hormone (PTH) examination of 1782 pg/mL (reference range: 15–65 pg/mL) and blood calcium of 3.54 mmol/L (reference range: 2.2–2.65 mmol/L). The diagnosis was hypercalcemia, hyperparathyroidism, and osteoporosis. She had a positive salmon calcitonin test sensitivity and was given symptomatic treatment with fluid replacement, hypocalcemic therapy (Ibandronate sodium monohydrate, Elcatonin), and diuresis. Despite 48 h of treatment, serum calcium levels rose to 3.9 mmol/L, prompting initiation of continuous renal replacement therapy (CRRT). Post-CRRT, calcium levels normalized to 2.475 mmol/L, with resolution of chest pain, CRRT was discontinued, and maintenance therapy with fluids, calcium-lowering agents, and diuretics resumed. However, calcium levels rebounded to 2.93 mmol/L within 48 h. Ultrasonography of the parathyroid glands showed that an uneven hypoechoic structure, measuring about 2.28 × 1.56 cm (
Figure 1(b) and (
c)), with fuzzy edges and irregular borders, was seen in the lower pole of the left lobe of the thyroid gland, and was characterized by abundant blood flow signals. Dual time-phase parathyroid examination showed that there were nodular shadows with slightly increased radioactivity distribution in the middle and lower part of the right lobe of the thyroid gland and in the lower part of the left lobe of the thyroid gland, which was considered to be the possibility of hyperfunctioning parathyroid tissue; and there was unevenly decreased bone density in the neck, bones of the thorax, and the upper part of the humerus bilaterally, with bone damage visible, which was considered to be the possibility of a brown tumor. Consultation with the Department of Thyroid Surgery was requested, Thyroid surgery consultation recommended parathyroidectomy.
Thyroid ultrasound indicates that multiple anechoic lesions are visible in the parenchyma of the left lobe, with the larger one located in the middle part, measuring approximately 0.27 cm in length, showing a regular shape, an aspect ratio of <1, and an intact margin. In the parenchyma of the middle-upper part of the right lobe, a solid hypoechoic lesion is visible, with a size of approximately 0.78 × 0.50 cm, presenting a regular shape, an aspect ratio of <1, and an intact margin; a mass of about 3.43 × 1.71 cm in size was detected in the dorsal part of the middle and lower part of the right lobe, with a regular morphology, aspect ratio <1, and clear borders (
Figure 2). The SPECT/CT fusion image of whole-body bone imaging showed active bone salt metabolism in the head, chest, spine, pelvis, and limbs, and the image of both kidneys was obviously diminished, which was in line with the imaging performance of bone imaging, and the possibility of metabolic bone disease caused by hyperparathyroidism was considered to be high, and the bone destructive shadow was shown, which was considered to be the possibility of brown tumors (
Figure 1(a)). After the patient’s state was stabilized, the patient and his family were fully communicated with the patient’s condition, and it was proposed to perform excision of parathyroid lesions, resection of thyroid goiter under general anesthesia.
After completing the relevant examinations, the patient’s blood calcium level was checked on the day of surgery, early in the morning: 3.45 mmol/L (reference range: 2.2–2.65 mmol/L); PTH was 1822 pg/mL (reference range: 15–65 pg/mL); procalcitonin was 4.7 pg/mL (reference range: <6 pg/mL). Intraoperative venous blood (I) samples were collected for rapid PTH monitoring (1510 pg/mL). No palpable nodules were identified in the left thyroid lobe. However, a nodule with a long diameter of approximately 0.8 cm was palpated on the surface of the right lobe of the thyroid, at the middle-upper part. An irregular mass measuring about 3.5 × 1.5 × 1.5 cm, partly cystic and separated from the thyroid gland, was palpable below the lower pole of the thyroid’s right lobe; a nodule measuring about 2.5 × 1.5 × 1.0 cm was identified inferior to the left lobe’s lower pole. The patient was diagnosed with hyperparathyroidism. In conjunction with the bilateral parathyroid imaging, the masses inferior to the lower poles of the thyroid gland were identified as hyperfunctional parathyroid tissue. Following excision of the left lower pole thyroid mass, venous blood (II) was drawn for PTH analysis (iPTH: 1601 pg/mL). Ten minutes postresection, a repeat PTH measurement (venous sample III) showed a decline to 899.1 pg/mL. Due to persistent elevation above the target range, the right lower pole mass was resected. A subsequent PTH assay (venous sample IV) at 10 min postexcision revealed a further reduction to 254.1 pg/mL. PTH decreased by more than 50% and returned to the normal range at 10-min postresection, confirming restoration of PTH homeostasis. Venous blood (V) drawn at 30-min interval revealed a PTH level of 135.9 pg/mL.
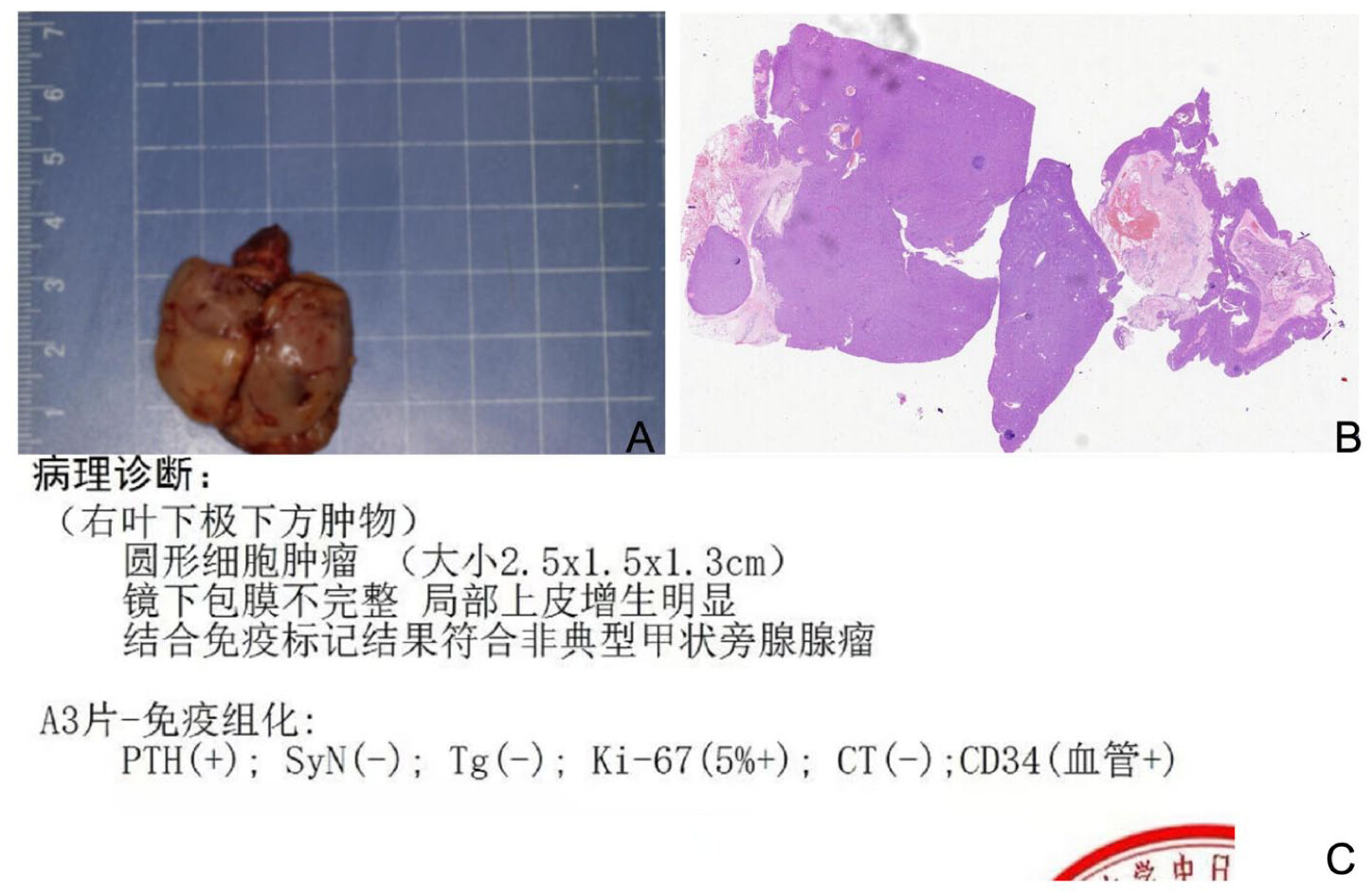
Table 1 describes the relationship between changes in intraoperative PTH levels and the resected masses. Excision of the nodule in the right lobe of the thyroid, along with adjacent normal glandular tissue and the masses located below the inferior poles of both lobes, was performed. The excised tissue was submitted for pathological examination. Intraoperative rapid pathological examination revealed the following: the left lobe lower pole mass showed features of a malignant tumor consistent with carcinoma; the right lobe lower pole mass was suggestive of a junctional tumor of follicular epithelial origin; and the right lobe portion showed nodular goiter with adenomatous hyperplasia of the thyroid gland. The gross specimens of the resected parathyroid carcinoma and parathyroid adenoma can be seen in
Figure 4. After communication with the patient’s family, the left side was treated as parathyroid carcinoma and performed resection of the thyroid left lobe and isthmus, along with central compartment lymph node dissection. Venous blood (VI) was collected 30 min postresection for PTH measurement, yielding a result of 34 pg/mL. (The relationship between intraoperative changes in PTH values and tumor resection is summarized in
Table 1.) Postoperative PTH levels were measured as 4.34 pg/mL at 24 h and 15.74 pg/mL at 48 h, with daily calcium supplementation of 4.8 g. Calcium levels remained within 1.99–2.58 mmol/L during the first three postoperative days.
Pathology report for paraffin-embedded tissue: (left lobe, inferior pole, inferior mass) parathyroid carcinoma (measuring 2.5 × 2 × 1.5 cm;
Figure 3(a)), immunohistochemistry results: PTH (+); CgA (+); SyX (−); Tg (−); Ki-67 (5%+); CT (−); CD34 (vascular +;
Figure 3(c)). (Right lobe, inferior pole, mass) Parathyroid adenoma (small, 2.5 × 1.5 × 1.3 cm;
Figure 4(a)), immunohistochemistry results: PTH (+); SyX (−); Tg (−); Ki-67 (5%+); CT (−); CD34 (vascular +;
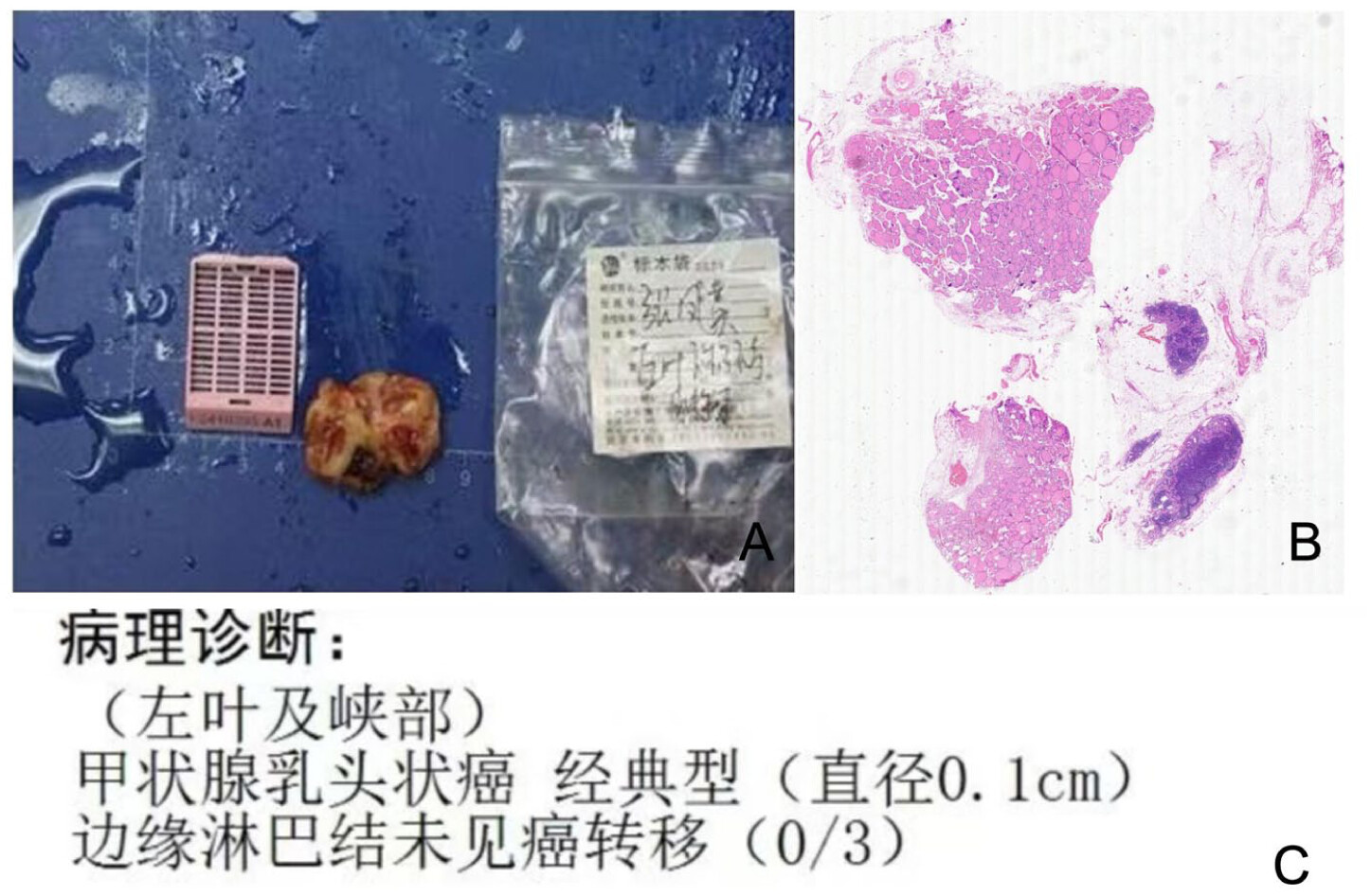
Figure 4(c)). (Left lobe and isthmus) Papillary thyroid carcinoma (PTC), classic variant (0.1 cm in diameter;
Figure 5); (right lobe portion) nodular goiter with thyroid adenomatous hyperplasia (
Figure 6); (central group) lymph nodes without evidence of cancer metastasis (0/10).
The patient was prescribed oral calcium supplementation at a dosage of 1.8 g daily postdischarge, with eugenol administered at a dose of 50 mcg/day to regulate endocrine function. At the 1-month follow-up in our hospital, dual-energy X-ray absorptiometry demonstrated improved bone mineral density: the lumbar spine T-score increased to −3.6 (preoperative: −3.8), while the left and right hip T-scores improved to −3.6 and −3.3, respectively (preoperative: −4.2 and −3.6). Biochemical analysis revealed serum calcium levels of 2.2 mmol/L, PTH of 15.53 pg/mL, and thyroid-stimulating hormone (TSH) of 3.63 mIU/L, all within normal reference ranges. Notably, eugenol dosage was titrated to maintain TSH levels within the target therapeutic window of 0.5–2 mIU/L. Subsequent evaluation at 6 months postoperatively showed sustained biochemical control, with PTH at 14.59 pg/mL and serum calcium at 2.34 mmol/L.
Discussion
A total of 22 cases of parathyroid carcinoma coexisting with thyroid cancer were documented between 1974 and 2024. Of these cases, only two involved the combination of parathyroid carcinoma and parathyroid adenoma with thyroid cancer. This case is the third reported case of parathyroid carcinoma combined with parathyroid adenoma and thyroid cancer. Of the 22 cases, 1 lacked relevant information, 15 patients were female and 6 were male, and the male-to-female ratio is 2:5. The age range of the patients’ onset of disease was between 21 and 89 years, and the present case was a female with an onset of disease at the age of 44 years. Clinically, parathyroid carcinoma is a rare endocrine malignancy. As it progresses, excessive PTH causes hypercalcemia and hyperparathyroidism. Unlike other malignancies, its symptoms stem not only from tumor infiltration but may also involve neuroendocrine dysfunction. Suggestive symptoms include age <55 years, PTH levels >10 times normal, hypercalcemia, severe skeletal/renal symptoms, palpable neck tumor, and recurrent laryngeal paralysis.
2 Of 22 reviewed patients, only 1 had normal preoperative blood calcium; the other 20 had varying hypercalcemia, and all had elevated PTH. The present patient also has hypercalcemia and elevated PTH.
The cause of parathyroid carcinoma remains poorly understood. It is generally accepted that it most likely arises directly rather than from benign adenoma transformation, given the distinct genetic alterations between adenomas and carcinomas.
4 Its association with thyroid cancer is extremely rare; when co-occurring, it is more likely linked to differentiated thyroid cancer (DTC), especially papillary type (PTC). While some consider this co-occurrence coincidental, others suggest factors like hypercalcemia, epithelial growth factor, or insulin-like growth factor may promote thyroid issues. Both sporadic parathyroid carcinoma and DTC have been linked to external radiation exposure.
4,5 Additionally, high glucagon-like peptide-1 receptor gene expression is thought to increase hyperparathyroidism risk.
6 The coexistence of multiple endocrine tumors, such as thyroid and parathyroid tumors, in a single patient is a complex clinical phenomenon whose underlying molecular mechanisms urgently require in-depth investigation. Multiple endocrine neoplasia (MEN) syndrome refers to a group of rare hereditary disorders characterized by the sequential or synchronous development of benign and malignant tumors in multiple endocrine glands.
7 The occurrence of such lesions is closely associated with genetic mutations, including MEN1 and RET gene mutations.
7 Although several subtypes of this syndrome have been clearly defined, the specific scenario of concurrent PTC, parathyroid adenoma, and parathyroid carcinoma in the same patient remains insufficiently elucidated. This rare manifestation may indicate the presence of novel genetic etiologies or represent sporadic, coincidental events. Recent studies have revealed genetic mutation correlations between thyroid carcinoma, parathyroid adenoma, and pheochromocytoma, and such molecular associations are more prominent in the context of MEN syndrome. Notably, the RET proto-oncogene has been identified as a key driver in the development and progression of medullary thyroid carcinoma and pheochromocytoma in MEN2
8: germline RET mutations are the primary cause of hereditary-related tumors, while somatic mutations are commonly observed in sporadic cases.
9 Overall, the genetic characteristics of these endocrine tumors are extremely complex, and the underlying mechanisms still require further clarification. Subsequent in-depth research in this field will provide important guidance for the clinical formulation of precise diagnosis and treatment strategies, the improvement of genetic counseling, and the exploration of pathogenic causes.
Hyperparathyroidism is usually caused by parathyroid adenoma or hyperplasia, whereas parathyroid carcinoma is extremely rare and easily confused with adenoma clinically. They can be differentiated via ultrasound, CT, MRI, PET/CT, 18F-choline-PET, and laboratory tests. Ultrasonographic features of parathyroid carcinoma include large size (>3 cm), irregular margins, aspect ratio >1, heterogeneous echogenicity, calcification, cystic-solid changes, abundant blood flow, and local tissue infiltration. Cystic-solid changes may relate to internal necrosis, liquefaction, or cystic transformation,
10 calcification may associate with heterogeneous proliferation and ossification of tumor cell membranes,
11 and carcinoma cells promote angiogenesis, causing abundant blood flow on ultrasound.
12 In our patient, parathyroid ultrasound revealed a lesion beneath the left lower pole of the thyroid gland. This lesion had blurred and irregular margins, calcifications, and abundant blood flow. In contrast, the lesion in the middle and lower lobes of the right thyroid had clear borders, a regular morphology, and an aspect ratio <1, resembling a thyroid nodule and potentially leading to a missed diagnosis. CT and MRI primarily detect local infiltration and distant metastasis. Malignant CT features include irregular margins, aspect ratio >1, calcification, peritumoral tissue infiltration, and mild enhancement. Benign features include short-axis to long-axis ratio <0.53 and high contrast enhancement (>56.6 HU in arterial phase; >59.5 HU in venous phase).
13 On MRI, parathyroid hyperplasia and adenomas usually show homogeneous signal, well-defined borders, and small size. In contrast, parathyroid carcinomas typically have heterogeneous signal, poorly defined borders, and large size.
14 In contrast, PET/CT can assess tumor local infiltration and distant metastasis, and is also suitable for identifying recurrence and detecting potential residual lesions post-treatment.
15 18F-choline PET is an advanced adjunctive technique for preoperative tumor localization. Notably, brown tumors, a pseudotumor due to overproduction of PTH, have 18F-choline PET images similar to parathyroid carcinomas, requiring careful differentiation to avoid confusion with metastases.
16 Parathyroid carcinoma should be highly suspected if blood calcium exceeds 12 mg/dL (3 mmol/L) and tumor diameter is over 3 cm. Additionally, Bae et al.’s
17 study noted that malignancy risk rises significantly when serum alkaline phosphatase exceeds 285 IU/L alongside tumor diameter >3 cm. A Korean study noted that PC patients had higher serum ALP levels than those with benign disease; PC lesions were usually over 3 cm in diameter and possibly palpable. Serum iPTH levels showed no statistically significant difference between PC and benign PHPT patients.
17 Our patient’s preoperative alkaline phosphatase level was 2194.67 U/L (reference range: 30–120 U/L), even though the diameter of the left parathyroid mass did not exceed 3 cm. Fine-needle aspiration biopsy (FNAB) is limited in distinguishing benign from malignant parathyroid tumors. It may even rupture the tumor envelope, risking cancer cell spread, so it is rarely used in diagnosing primary parathyroid tumors.
18 However, FNAB can serve as a preoperative diagnostic criterion for thyroid cancer. Immunohistochemistry also enhances diagnostic accuracy for parathyroid cancer. Typically, PTH (+), CgA (+), Tg (−), and CT (−) confirm parathyroid tissue.
19 GATA-3 is a reliable marker of parathyroid differentiation, aiding in the differential diagnosis of parathyroid tumors.
20 The most common biomarkers for malignant diagnosis are Ki-67 and parafibromin; a Ki-67 proliferation index >5% in parathyroid tumors suggests malignancy.
21–23 In our patient, postoperative immunohistochemistry (PTH (+); CgA (+); SyX (−); Tg (−); Ki-67 (5%+); CT (−); CD34 (vascular +)) suggested parathyroid carcinoma. However, postoperative pathologic diagnosis remains the gold standard for confirming it, with other investigations as adjuncts for preoperative identification.
Acute kidney injury is relatively common in PHPT patients, while hypercalcemic crisis affects only 1%—2% of them.
24 PHPT and malignancy-associated hypercalcemia account for over 90% of common hypercalcemia causes. Other less common ones include endocrine disorders such as hyperthyroidism, acromegaly, pheochromocytoma, and some adrenocortical insufficiencies. Hypercalcemic patients are often dehydrated, so 0.9% sodium chloride injection should correct dehydration before drug therapy. Meanwhile, loop diuretics, calcimimetics, and antiresorptive agents (e.g., bisphosphonates or denosumab) should be given to effectively control life-threatening hypercalcemia.
25 Calcitonin, the most commonly used clinical calcium-lowering drug, acts rapidly and can be combined with hydration as initial adjunctive therapy. Bisphosphonates, another common clinical calcium-lowering class, have a long duration but slower onset. Cinacalcet, a calcimimetic, controls blood calcium and reduces PTH by modulating calcium-sensing receptors.
25 For nonsurgical patients, calcimimetics are currently the most effective for controlling hypercalcemia. In hypercalcemic crisis, besides the above timely conservative measures, studies show hemodialysis with low- or no-calcium dialysate can rapidly correct severe hypercalcemia, making it a first-choice treatment.
26 Our patient was admitted with hypercalcemia (serum calcium level: 3.54 mmol/L) and no evidence of acute kidney injury. The patient’s salmon calcitonin test yielded a positive result; however, symptomatic interventions including fluid rehydration, ibandronate sodium monohydrate, elcatonin, and diuretics failed to control the condition, with serum calcium levels continuing to rise to 3.9 mmol/L. Following 2 days of CRRT, the serum calcium level decreased to 2.475 mmol/L, and CRRT was therefore discontinued. Despite the continuation of rehydration, calcitonin administration, diuretic therapy, and other symptomatic measures, serum calcium rebounded to 2.93 mmol/L after an additional 2 days. These findings collectively indicate that conservative treatment was ineffective in managing this patient’s hypercalcemia. When conservative treatment fails, surgery is the first-line option for hyperparathyroidism. Surgical indications include symptomatic hyperparathyroidism; asymptomatic PHPT with any of the following: hypercalcemia (0.25 mmol/L above upper normal limit), renal impairment (creatinine clearance <60 mL/min), bone mineral density <2.5 quartiles below peak mass at any site (
t-value <−2.5), and/or fragility fractures, age <50 years, inability to undergo routine follow-up, or well-localized lesions without surgical contraindications. Most studies recommend that initial surgery resect the tumor, ipsilateral thyroid lobe, isthmus, and adjacent structures to better control local disease and improve long-term survival. Intraoperative tumor envelope rupture should be avoided to prevent tumor cell spillage.
27 Parathyroid adenomas are usually treated by removing the tumor. If the adenoma is within the thyroid, the thyroid and tumor should be removed together, with assessment of the other parathyroid glands. For preoperatively well-localized adenomas, only the tumor needs removal during surgery without exploring the contralateral parathyroids.
27 The standard surgical approaches for PTC include unilateral lobectomy (plus isthmus) and total/subtotal thyroidectomy. Unilateral lobectomy (plus isthmus) is recommended for PTC with lesions <1 cm and no high-risk factors, with specific approaches selected individually based on multiple factors. Based on the preoperative diagnosis and established surgical indications, we resected the masses located inferior to the lower poles of both the left and right thyroid lobes. Intraoperative PTH levels were continuously monitored to evaluate parathyroid gland function. Intraoperative frozen section pathology confirmed malignancy of the left lower pole mass; following detailed discussion and informed consent from the patient’s family, we proceeded with oncological resection on the left side, which included complete excision of the left thyroid lobe, thyroid isthmus, and central neck compartment lymph node dissection.
Although postoperative PTH and calcium levels normalize or even drop below normal, this does not indicate full recovery; parathyroid cancer has a high recurrence rate even after radical resection. Median overall survival from diagnosis is 14.3 years (range 10.5–25.7 years). Of 22 reviewed parathyroid cancer patients, only 11 had follow-up records: 2 recurred, 1 died, and 8 maintained normal calcium and PTH levels. The current patient, with 6 months of postoperative follow-up, has maintained normal blood calcium and PTH levels. Factors increasing mortality include lymph node or distant metastasis, recurrence frequency, high blood calcium at recurrence, and extensive use of calcium-lowering drugs. Conversely, factors unrelated to mortality include age, race, tumor size, time to first recurrence, and initial surgical extent.
28 Parathyroid carcinoma most commonly metastasizes to cervical lymph nodes, lungs, and liver, with prognosis varying significantly by site. Cervical lymph node metastases occur in up to 19% of cases, more commonly when tumors exceed 3 cm.
29 Currently, our patient’s primary tumor is confined to the parathyroid glands, without lymph node involvement or distant metastasis. Thus, early diagnosis and complete resection of the lesion are crucial for improving prognosis. For metastatic parathyroid carcinoma, main treatments include dacarbazine- and anthracycline-based chemotherapy, plus targeted anti-angiogenic therapies: sorafenib, cabozantinib, and lenvatinib.
30 Additionally, immunotherapy is a promising avenue: two studies on PC immunogenicity reported PDL1 expression in 22% and high mutational load in 18% of parathyroid cancer cases.
31,32







{kind=link}